For each batch of thiolated HA, the degree of thiolation should be verified using H1-NMR or an Ellman's test. HA modification using the procedure described here consistently generates ~5% thiolation (defined as the molar ratio of thiols to HA disaccharides) (Figure 1).
Setting up this new culture platform will require each laboratory to perform rigorous testing to ensure good culture viability prior to implementing large-scale experiments. Our 80 µL hydrogels with a seeding density 500,000 cells/mL (40,000 cells/gel) consistently result in proliferation rates that are comparable to, or better than, gliomasphere cultures (Figure 2A-2C). As HA/PEG hydrogels are optically transparent, cell behaviors can be observed directly in live, 3D cultures using phase contrast or fluorescence microscopy. Figure 3A shows that, 4 days after encapsulation, GBM cells in RGD-containing hydrogels exhibit an invasive phenotype, while cells cultured in hydrogel using PEG-MAL-CYS controls have a spherical morphology.
As is typical with xenograft models where researchers must wait for days to months until implanted tumors reach progressive growth24,32, patient-derived cells also take time to adjust to a new culture environment. Thus, we recommend culturing 4-8 days before commencing experiments, such as drug treatment, to ensure most cells have entered the exponential growth phase. Beyond drug treatments, reagents like soluble cyclo-RGD, which competitively disrupt cell interactions with RGD in hydrogels, can be added to culture medium (Figure 3A).
Our hydrogel system is compatible with many common methods for investigating glioma cell biology. Using bioluminescence imaging, which is commonly used to monitor rodent xenograft tumors, relative numbers of viable cells can be observed in hydrogel cultures during the course of treatment. Figure 3B provides an example of this method, where effects of erlotinib treatment on hydrogel-cultured GBM cells were evaluated over 6 days. In general, hydrogel cultures can be treated as tissue samples when preparing lysates for Western blot or PCR. Western blot analysis of cleaved poly ADP polymerase indicates that relative degree of apoptosis in treated CD44 knockdown cells is higher than wildtype GBM cells cultured in 0.5% HA hydrogels (Figure 3C). Similarly, single cell suspensions can be prepared from hydrogel cultures for analysis via flow cytometry using standard protocols for liberating single cells from intact tissues (Figure 2). In addition to cell features, cryosections of hydrogel-based, 3D cultures preserve ECM deposited by cultured cells. For example, deposition patterns of type IV collagen shift upon erlotinib treatment of hydrogel cultures (Figure 3D).
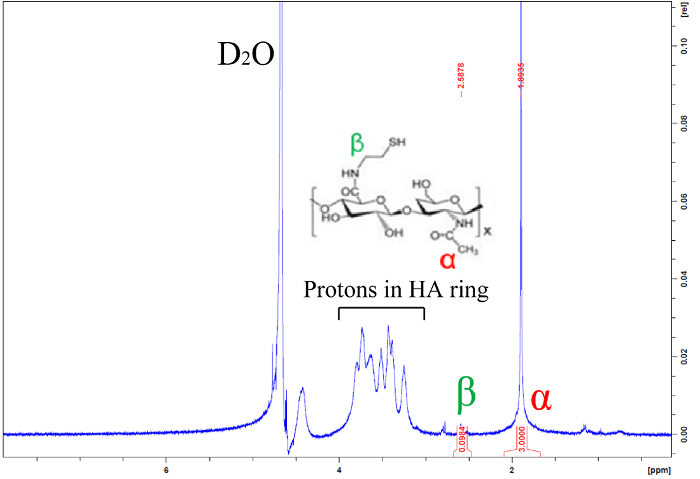
Figure 1: Representative H1-NMR spectrum of thiolated hyaluronic acid. Integrated peaks indicate that approximately 5% of HA glucuronic acid groups have been modified with a thiol. Please click here to view a larger version of this figure.
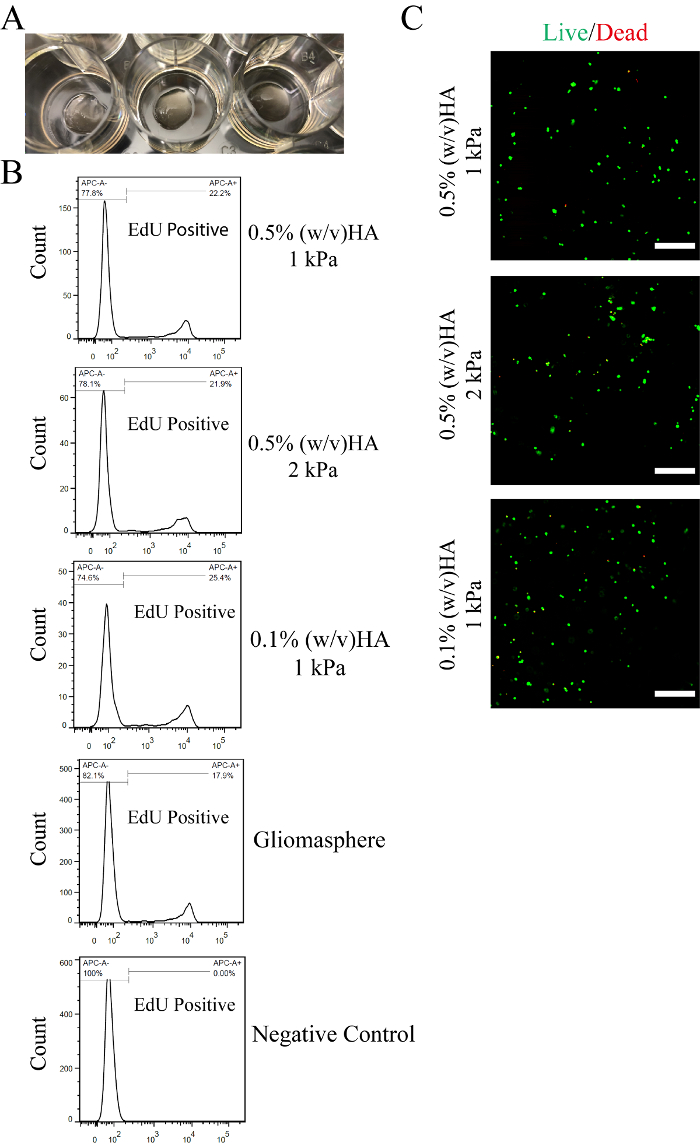
Figure 2: Proliferation of hydrogel-encapsulated cells. A) Example images of fabricated 80 µL gels in 12-well plates. After crosslinking, hydrogels were swollen in cell culture medium. B) Representative results of proliferation rate measured using flow cytometry. GBM cells (HK301) were incubated with 1 µM EdU (5-ethynyl-2′-deoxyuridine) for 2.5 h on the fourth day after encapsulation or passaging. A click-reaction was used to conjugate fluorescent dye to incorporated EdU, as detailed in Xiao et al. 2017.3 Negative controls, where no EdU was added to cultures, were included. C) Representative confocal microscopy images of live (green) and dead (red) cells 24 hours after hydrogel cultures of GBM cells (HK157) were established. Scale bars = 200 µm. Please click here to view a larger version of this figure.
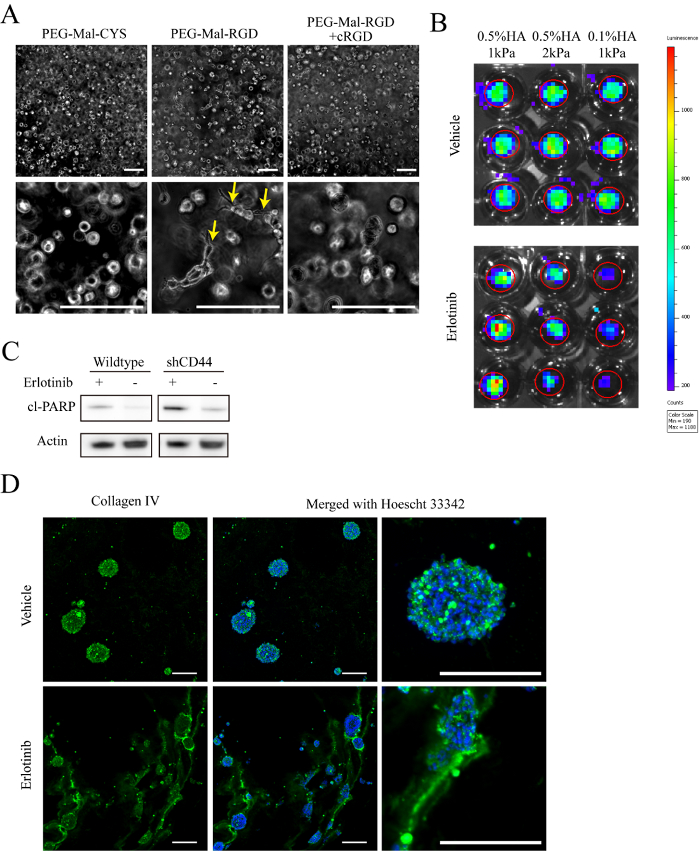
Figure 3: Characterization. A) Representative phase contrast images of 0.5% (w/v) HA hydrogel-cultured cells under varying conditions for 8 days after encapsulation. Arrows indicate cells with an invasive morphology. B) Representative images of bioluminescence signal measured after 15 days of treatment with 1 µM erlotinib or vehicle (DMSO). 1 mM D-luciferin was added to cell culture medium 1 h prior to imaging. Cells were transduced with lentivirus encoding for constitutive expression of firefly luciferase prior to encapsulation. C) Representative immune-blot images analyzing cleaved poly ADP polymerase (cl-PARP) expression in GBM cells (HK301) cultured in hydrogels with 0.5% (w/v) HA and 1 kPa compressive modulus. All cropped images shown were from the same blot. D) Representative staining of collagen IV (green) and Hoechst 33342 (blue) in hydrogel-cultured HK301 cells 12 days after treatment with 1 µm erlotinib or vehicle (DMSO). Scale bar = 200 µm. Please click here to view a larger version of this figure.
| Gel Type | Part A (40 µL each) | Part B (40 µL each) | ||||
| 4Arm-PEG-MAL (50mg/mL) | Cysteine or RGD (2.81mM) | PBS (pH 7.4) | 4Arm-PEG-thiol (50mg/mL) | PBS (pH 7.4) | HA-S (13.3mg/mL) | |
| 0.5% (w/v) HA 1kPa | 10 µL | 4.00 µL | 26 µL | 1.00 µL | 9.00 µL | 30.0 µL |
| 0.5% (w/v) HA 2kPa | 20.0 µL | 4.00 µL | 16.0 µL | 8.00 µL | 2.00 µL | 30.0 µL |
| 0.1% (w/v) HA 1kPa | 10 µL | 4.00 µL | 26 µL | 8.00 µL | 26.0 µL | 6.00 µL |
Table 1. Hydrogel formulations yielding independent control of HA concentration and mechanical properties.
