Strategies for rAaeDV construction
A defective rAaeDV vector was generated to express the DsRed gene in mosquito larvae. The resulting plasmid contained a NS1-DsRed fusion protein cassette with the VP protein deleted (Figure 1A). rAaeDV plasmids containing expression constructs for miRNA, miRNA sponge, shRNA and amiRNA were designed (shown in Figure 1B). For example, a rAaeDV vector was generated to overexpress aal-let-7 in mosquito larvae. The resulting plasmid contained an intronic pre-aal-let-7 cassette in the viral NS1 exon (Figure 1B). A rAaeDV vector containing an intronic aal-let-7 sponge expression cassette in the viral NS1 exon was generated to knock down aal-let-7 expression in mosquito larvae (Figure 1B and Figure 2B). A rAaeDV shRNA vector and a rAaeDV amiRNA vector containing intronic shRNA and pre-amiRNA expression cassettes, respectively, were used to knock down V-ATPase mRNA in mosquito larvae (Figure 2C).
Calculating the rAaeDV titers based on a standard curve that generated with the linear regression analysis
The y-axis represents the Log10 value of standard DNA molecular concentration, and the x-axis indicates the CT value. The CT numbers (x-axis) and Log10 (concentration) values (y-axis) displayed a good correlation (R2 = 0.9988) and meet the equation y = – 0.288x + 13.87 (Figure 3). Calculate the rAaeDV titers of samples using the linear equation (Table 1). Using the method described in the protocol (step 6.2 and Table 1), the high titers of rAaeDV (4 mL, >7.65 x 1010 particles/mL) were harvested. Recombinant viruses (VrepUCA-7, VrepUCA-7s, VrepUCA-amiVTP, VrepUCA-shVTP, VrepUCA941-1, VrepUCA-941-1s, and VrepUCA-shct) were generated by transfecting the corresponding infection clones pUCA-7, pUCA-7s, pUCA-amiVTP, pUCA941-1, pUCA941-1s, pUCA-shct into C6/36 cells (sections 3, 4 and 5).
Determining the miRNA overexpression and knockdown efficiency of rAaeDV vectors
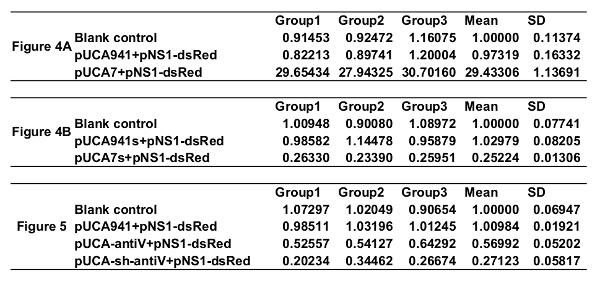
In the example, 100 1st instar larvae were treated with equal amounts (1.00 x 1010 particles/mL) of rAaeDV containing the intronic pre-let-7 expression vector, the intronic let-7 sponge expression vector, the human-specific pre-miR-941-1 expression vector or the miR-941-1 sponge expression vector by adding the virus into the water body of the larval habitat. Five days after infection, the expression of aal-let-7 was determined by qPCR (Figure 4, n = 3; Supplemental Table 2). The let-7 levels that are displayed as the mean ± SD of three biological replicates in the larvae, were substantially increased or decreased by the pre-let-7-expressing rAaeDV (overexpressed by 2,843.31 ± 80.72%, p <0.0001, t-test) and the let-7-sponge-expressing rAaeDV (downregulated by 74.78 ± 1.60%, p <0.0001, t-test), respectively.
Monitoring the V-ATPase mRNA knockdown efficiency of rAaeDV vectors
A total of 100 1st instar larvae were treated with equal amounts (1.00 x 1010 particles/mL) of intronic amiRNA-expressing rAaeDV, intronic shRNA-expressing rAaeDV, human-specific pre-miR-941-1-expressing rAaeDV orscramble shRNA-expressing rAaeDV by adding the virus into the water body of the larval habitat. Five days after infection, the expression of V-ATPase mRNA was determined by qPCR (Figure 5, n = 3; Supplemental Table 2). The V-ATPase level displayed as the mean ± SD of three biological replicates in the larvae was substantially reduced by amiRNA-expressing rAaeDV (downregulated by 43.01 ±6.37%, p = 0.0011, t-test) and intronic shRNA-expressing rAaeDV (downregulated by 72.88 ± 5.74%, p = 0.0001, t-test).
Figure 1: Schematic organization of the recombinant AaeDV plasmids. (A) The pNS and pVP viral promoters drive the expression of the NS1/NS2 genes and VP genes, respectively. In the p7NS1-DsRed plasmid, the DsRed gene was fused to the NS1 gene. (B) pUCA-7, pUCA-7s, pUCA-941-1, pUCA-941-1s, pUCA-shct, pUCA-shVTP and pUCA-amiVTP contain artificial introns, including the aal-let-7, aal-let-7 sponge, hsa-miR-941-1, hsa-miR-941-1 sponge, scrambled shRNA, V-ATPase shRNA and artificial miRNA, respectively, which were cloned into the HpaI site of the NS1 gene. The artificial intron is shown flanked by a splice donor (DS) and an acceptor site (AS) and contains a branch-point domain (BrP), a poly-pyrimidine tract (PPT) and pre-miRNA. The miRNA sponge or artificial miRNA sequence is inserted inside the intron between the 5-splice site and the BrP. Please click here to view a larger version of this figure.
Figure 2: Biogenesis of artificial intronic microRNA (miRNA) and strategy for generating miRNA sponges and artificial miRNAs. (A) Intronic miRNA is co-transcribed within a precursor messenger RNA (pre-mRNA) of NS1, which will be driven by the pNS1 promoter and cleaved out of the pre-mRNA by RNA splicing. Although the exons are ligated to form a mature messenger RNA (mRNA) for NS1 protein synthesis, the spliced intron with the pre-miRNA is further processed into mature miRNA by Dicer. (B) Strategy for generating the anti-let-7 and anti-miR-941-1 constructs. Alignment of anti-let-7 and anti-miR-941-1 sequences with aal-let-7 and hsa-miR-941-1, respectively. Complete matching of the seed regions with the anti-miRNA sequence and tail regions is shown. Both let-7 and miR-941-1 sponges contain three repeat antisense constructs (red letters) that can bind to aal-let-7 and hsa-miR-941-1, respectively. (C) Sequences and predicted precursor structures for the miRNA-based artificial miRNAs and shRNA used in this study. The mature artificial miRNAs and shRNA are shown in red, and their related target mRNA sequences are in blue. Please click here to view a larger version of this figure.
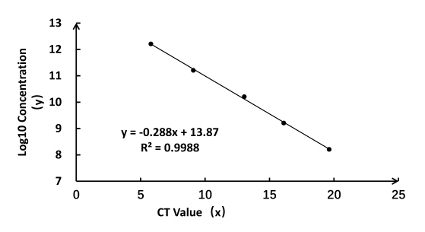
Figure 3: Standard curve for rAaeDV titration and the calculated concentrations of MDVs. (A) Real-time qPCR was performed with a pUCA standard that was diluted by 10 times the magnification ratio, and the CT value was measured on an ABI 7500 as x. Copy number was calculated according to step 6.2 and 6.3 with the following formula: copy number (y) = -0.288x + 10.87; R2 = 0.9988. Please click here to view a larger version of this figure.
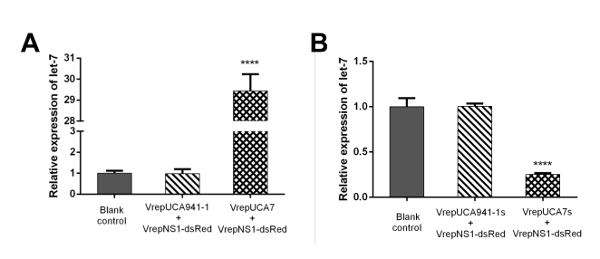
Figure 4: Recombinant MDV-mediated aal-let-7 overexpression and knockdown in Ae. albopictus larvae. (A) Analysis of endogenous miRNA expression in larvae after infection with recombinant virus containing the aal-let-7 expression cassette (left panel). (B) The efficiency of the anti-let-7 construct was determined in larvae (right panel). The has-miR-941-1 and has-941-1 sponge (negative control) overexpressed and downregulated the mature let-7 miRNAs, respectively, compared to the basal levels observed in the control group. miRNA abundance was normalized to 5S rRNA, and the data are displayed as the mean ± SD of three biological replicates. T-tests were performed at different levels of significance. The asterisks indicate statistically significant differences between the infected and corresponding mock-treated larvae (four asterisks, p <0.0001). Please click here to view a larger version of this figure.
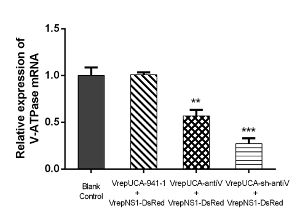
Figure 5: Analysis of V-ATPase mRNA expression after infection with recombinant virus containing shRNA and amiRNA cassettes. mRNA abundance was normalized to aalrpS7. Error bars represent the standard deviation of the 2−ΔΔCT values for V-ATPase mRNA expression in the Ae. albopictus larvae as evaluated by real-time RT-PCR (**, p <0.01; ***, p <0.001). Please click here to view a larger version of this figure.
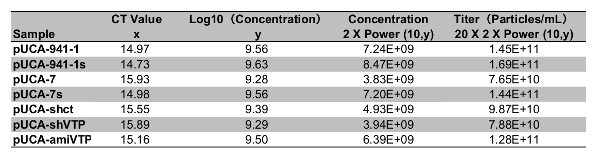
Table 1: The recombinant MDV CT value determined by qPCR corresponds to the x value in the formula, which is used to calculate the y value. The viral genome copy number in each 1 µL sample was obtained by the function power (10, y), which was ultimately multiplied by 40, converting the value to the virus concentration of virus suspension.
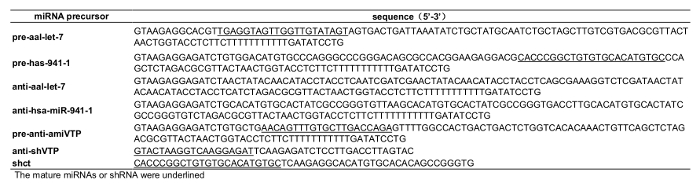
Supplemental Table 1: Sequences of miRNA precursor.
Supplemental Table 2: qPCR data.
