Quelle: Susannah C. Shissler1, Tonya J. Webb1
1 Institut für Mikrobiologie und Immunologie, University of Maryland, Baltimore, MD 21201
Immunpräzipitation (IP, auch bekannt als “Pull-down”-Assay) ist eine weit verbreitete Technik, die Anwendungen in einer Vielzahl von Bereichen hat. Erstmals 1984 konzipiert, wurde es 1988 verfeinert (1, 2). Das grundlegende Ziel der IP ist die Reinigung und Isolierung eines bestimmten Proteins mit einem Antikörper gegen dieses Protein. Das Wort “Immuno” bezieht sich auf die Verwendung eines Antikörpers, während das Wort “Ausfällung” sich auf das Abziehen einer bestimmten Substanz aus einer Lösung bezieht. Das Zielprotein kann endogene oder rekombinant ewanant sein. Die meisten rekombinanten Proteine haben ein Epitop-Tag (d. h. myc oder Flag), das an ihnen angebracht ist, um die nachfolgende Reinigung zu vereinfachen. In der Regel ist es einfacher, rekombinante Protein-IP zu optimieren, da die Antikörper gegen rekombinante Epitop-Tags sehr stark und wirksam sind. Antikörper gegen endogene Proteine haben eine extrem variable Wirksamkeit , was es viel schwieriger macht, diese IPs zu optimieren. Ein notwendiger Schritt nach der Immunpräzipitation ist die Überprüfung der Reinigung. Das isolierte Protein wird mit SDS-PAGE aufgelöst und anschließend durch westliche Flecken auf Reinheit untersucht (Abbildung 1). Eine wichtige Kontrolle ist die Verwendung eines anderen Antikörpers während des Western Blot, um den Abziehdes des richtigen Proteins zu überprüfen. Die Kombination von IP mit nachfolgenden Techniken ist ein leistungsstarkes Analysetool. Das Ziel nach der Reinigung kann die Charakterisierung des Proteins selbst durch NMR, Massenspektrometrie und In-vitro-Assays oder die Analyse der interagierenden Partner des Proteins (z. B. Protein, DNA, RNA) (3, 4, 5) sein.
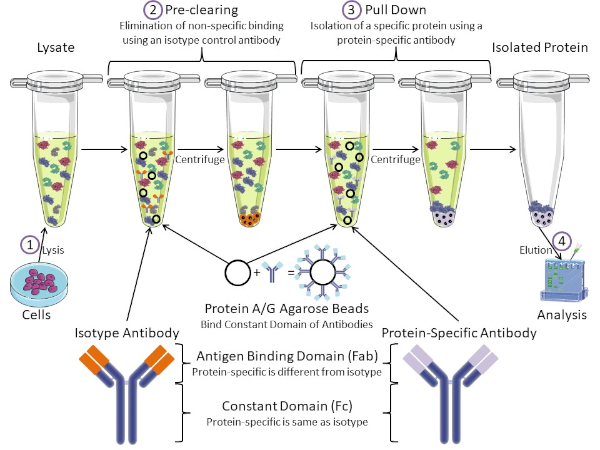
Abbildung 1: Überblick über das Immunpräzipitungsverfahren. Immunpräzipitation ist die Isolierung eines bestimmten Proteins mit einem Antikörper. Nach der Produktion von Lysat aus Zellen, gibt es zwei wichtige Schritte – Vor-Clearing und ziehen nach unten. Während des Vorklärungsschritts werden die Zelllysate von Proteinen vorgereinigt, die sich nicht spezifisch mit einem Isotyp-Kontrollantikörper an Antikörper binden. Im Pull-Down-Schritt wird das Zielprotein mit einem proteinspezifischen Antikörper nach unten gezogen. Das isolierte Protein wird dann durch Western Blot analysiert. Isotyp-Antikörper und proteinspezifische Antikörper haben die gleiche konstante Domäne, aber unterschiedliche Antigen-Bindungsdomänen. Ein wichtiger Bestandteil dieses Protokolls sind Protein-A/G-Agarose-Perlen, die die konstante Domäne von Antikörpern binden und eine Immunpräzipation des Zielproteins ermöglichen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Antikörper sind der Schlüsselbestandteil einer Immunpräzipitation, die sie von anderen Formen der Proteinreinigung (d. h. Nickelaffinitätssäulenreinigung) unterscheidet. Antikörper sind Moleküle von B-Zellen, die bestimmte Proteinepitope erkennen können. Antikörper haben zwei Domänen: Konstante (Fc) und Antigenbindung (Fab) (Abbildung 1). Die konstante Domäne identifiziert den Typ des Antikörpers und diktiert die Funktion in vivo. In der Regel sind die konstanten Domänen von Antikörpern, die für IP verwendet werden, Maus, Ratte oder Kaninchen IgG. Der Antigenbindungsanteil des Antikörpers erkennt ein spezifisches Epitop eines bestimmten Proteins. Antikörper können Epitope an gefalteten Proteinen erkennen, die möglicherweise nicht existieren, wenn das Protein denaturiert ist und umgekehrt. Daher hängt die Verfügbarkeit des Epitops von der Proteinfaltung ab – die Identifizierung eines wichtigen Faktors, der bei der Wahl von Antikörpern und Bedingungen für IP zu berücksichtigen ist.
Sowohl prokaryotische als auch eukaryotische Systeme haben Antikörper-bindende Proteine. In eukaryotischen Systemen, der Zweck ist Immunschutz vor Bakterien, während in prokaryotischen Systemen, der Zweck ist der Schutz vor dem Immunsystem. Antikörperbindende Proteine beeinflussen die IP-Methodik auf zwei Arten. Erstens gibt es einen notwendigen Vorklärungsschritt (Abbildung 1), um das Lysat von Proteinen zu befreien, die Antikörper binden – wodurch die unspezifische Bindung im Endprodukt reduziert wird. In diesem Schritt wird ein Isotyp-Antikörper verwendet, der die gleiche konstante Domäne wie eine andere Antikörperbindungsdomäne als Ihr proteinspezifischer Antikörper hat. Bakterielle Antikörper-bindende Proteine sind die zweite Schlüsselkomponente dieser Methode. Nachdem der proteinspezifische Antikörper das Zielprotein bindet, muss der Antikörper: Proteinkomplex heruntergezogen werden (Abbildung 1). Proteine A, G und L sind bakterielle Proteine, die die konstante Domäne von Antikörpern binden. Während Bakterien dies verwenden, um das Immunsystem zu untergraben, Forscher haben dieses System für eine einfache Antikörperreinigung kooptiert, und es wird sowohl während der Vor-Clearing-und Pull-down-Schritte verwendet. Diese Proteine haben unterschiedliche Bindungsaffinitäten für verschiedene Arten und verschiedene konstante Domänensubtypen – ein weiterer Faktor, der bei der Auswahl der Bedingungen für IP zu berücksichtigen ist. Viele Unternehmen verkaufen Protein A/G-markierte Agaroseperlen (Abbildung 1), vorgefertigte Spinsäulen oder Harze, um Säulen herzustellen. Im Allgemeinen werden Perlen und Spinsäulen für kleinere Probengrößen verwendet, während Harze für die Massenreinigung verwendet werden.
In dieser Übungsübung zeigen wir, wie das endogene Protein c-myc aus primären murinen Thymosyten mit Protein A/G Plus Agaroseperlen auf basisiger Immunpräzipitationstechnik gereinigt werden kann. Das Protokoll beginnt mit der Zelllysatzubereitung und endet mit der Überprüfung des erfolgreichen Protein-Pull-Downs mittels Western-Blot-Analyse.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.
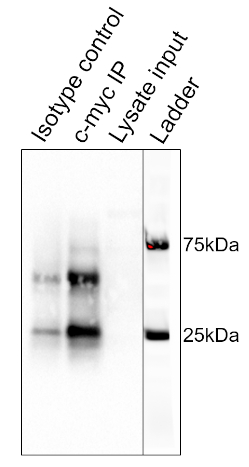
Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.