Fonte: Susannah C. Shissler1, Tonya J. Webb1
1 Departamento de Microbiologia e Imunologia, Universidade de Maryland, Baltimore, MD 21201
Imunoprecipitação (IP, também conhecido como ensaio ‘pull-down’) é uma técnica amplamente utilizada que tem aplicações em uma variedade de campos. Concebido pela primeira vez em 1984, foi refinado em 1988 (1, 2). O objetivo fundamental do IP é a purificação e isolamento de uma proteína específica usando um anticorpo contra essa proteína. A palavra “imuno” refere-se ao uso de um anticorpo, enquanto a palavra “precipitação” refere-se a retirar uma substância específica de uma solução. A proteína alvo pode ser endógena ou recombinante. A maioria das proteínas recombinantes tem uma tag de epítope (ou seja, myc ou flag) anexada a elas para simplificar a purificação subsequente. Normalmente, é mais fácil otimizar a proteína recombinante IP porque os anticorpos contra tags de epitope recombinantes são muito fortes e eficazes. Anticorpos contra proteínas endógenas têm eficácia extremamente variável – tornando muito mais difícil otimizar esses IPs. Um passo necessário após a imunoprecipitação é a verificação da purificação. A proteína isolada é resolvida usando SDS-PAGE e posteriormente sondada para pureza por manchas ocidentais (Figura 1). Um controle importante é o uso de um anticorpo diferente durante a mancha ocidental para verificar a retirada da proteína correta. A combinação de IP com técnicas subsequentes é uma poderosa ferramenta de análise. O objetivo após a purificação pode ser a caracterização da proteína em si por NMR, espectrometria de massa e ensaios in vitro, ou análise dos parceiros interativos da proteína (ou seja, proteína, DNA, RNA) (3, 4, 5).
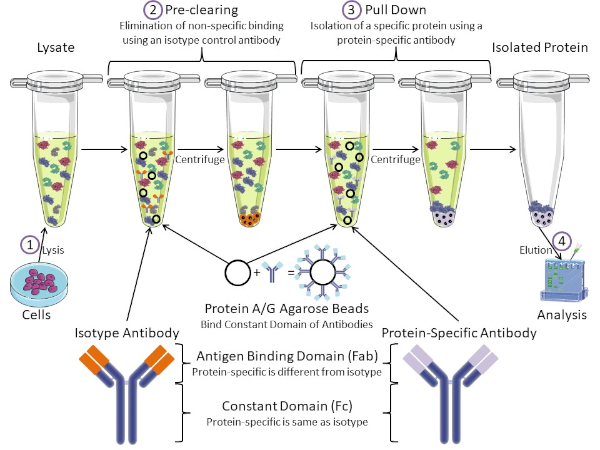
Figura 1: Visão geral do Procedimento de Imunoprecipitação. Imunoprecipitação é o isolamento de uma proteína específica usando um anticorpo. Após a produção de lise das células, há dois passos principais: pré-limpar e puxar para baixo. Durante a etapa de pré-limpeza, os lysatos celulares são pré-eliminados de proteínas que se ligam a anticorpos não especificamente usando um anticorpo de controle de isótipo. Em pull down step, a proteína alvo é puxada para baixo usando um anticorpo específico de proteína. A proteína isolada é então analisada pela mancha ocidental. Anticorpos isótipos e anticorpos específicos de proteína têm o mesmo domínio constante, mas diferentes domínios de ligação de antígeno. Um componente-chave deste protocolo são as contas de agarose da Proteína A/G que ligam o domínio constante dos anticorpos, permitindo a imunoprecipitação da proteína alvo. Clique aqui para ver uma versão maior desta figura.
Os anticorpos são o componente-chave de uma imunoprecipitação que o diferencia de outras formas de purificação de proteínas (ou seja, purificação da coluna de afinidade de níquel). Anticorpos são moléculas feitas por células B que podem reconhecer epítopos proteicos específicos. Os anticorpos possuem dois domínios: ligação constante (Fc) e antígeno (Fab) (Figura 1). O domínio constante identifica o tipo de anticorpo e dita a função in vivo. Normalmente, os domínios constantes dos anticorpos usados para IP são mouse, rat ou rabbit IgG. A porção de ligação de antígeno do anticorpo reconhece um epítope específico de uma proteína específica. Anticorpos podem reconhecer epítopos em proteínas dobradas que podem não existir quando a proteína é desnaturada e vice-versa. Portanto, a disponibilidade do epítope depende da dobra de proteínas – identificando um fator importante a considerar na escolha de anticorpos e condições para IP.
Ambos os sistemas procarióticos e eucarióticos têm proteínas de ligação de anticorpos. Nos sistemas eucarióticos, o objetivo é a proteção imune contra bactérias enquanto em sistemas procarióticos, o objetivo é a proteção do sistema imunológico. Proteínas de ligação de anticorpos afetam a metodologia IP de duas maneiras. Em primeiro lugar, há uma etapa de pré-compensação necessária (Figura 1) para livrar o lise de proteínas que ligam anticorpos – reduzindo assim a vinculação não específica no produto final. Esta etapa usa um anticorpo isótipo que tem o mesmo domínio constante que, mas um domínio de ligação de anticorpos diferente do seu anticorpo específico para proteínas. Proteínas bacterianas de ligação de anticorpos são o segundo componente-chave deste método. Depois que o anticorpo específico da proteína liga a proteína alvo, o anticorpo: complexo proteico deve ser puxado para baixo (Figura 1). Proteínas A, G e L são proteínas bacterianas que ligam o domínio constante dos anticorpos. Embora as bactérias usem isso para subverter o sistema imunológico, os pesquisadores têm cooptado este sistema para uma fácil purificação de anticorpos, e ele é usado durante os passos pré-clareira e pull-down. Essas proteínas têm diferentes afinidades de ligação para diferentes espécies e diferentes subtipos de domínio constantes – outro fator a considerar ao escolher condições para IP. Muitas empresas vendem contas de agarose rotuladas protein A/G (Figura 1), colunas de spin pré-feitas ou resinas para fazer colunas. Em geral, contas e colunas de spin são usadas para tamanhos amostrais menores, enquanto resinas são usadas para purificação a granel.
Neste exercício de laboratório, demonstramos como purificar a proteína endógena c-myc, dos timócitos murinas primários, utilizando contas de proteína A/G Plus baseadas em imunoprecipitação básica. O protocolo começa a partir da preparação de lisecelulares e termina com a verificação de retirada de proteínas bem sucedidas usando a análise de manchas ocidentais.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.
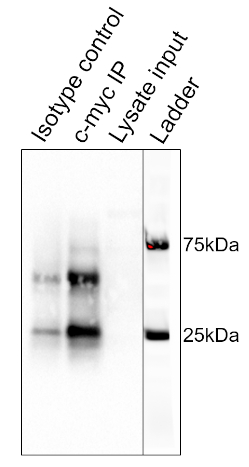
Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.