Fuente: Susannah C. Shissler1, Tonya J. Webb1
1 Departamento de Microbiología e Inmunología, Universidad de Maryland, Baltimore, MD 21201
La inmunoprecipitación (IP, también conocida como ensayo “pull-down”) es una técnica ampliamente utilizada que tiene aplicaciones en una variedad de campos. Concebido por primera vez en 1984, fue refinado en 1988 (1, 2). El objetivo fundamental de la P.I. es la purificación y el aislamiento de una proteína específica utilizando un anticuerpo contra esa proteína. La palabra “inmuno” se refiere al uso de un anticuerpo, mientras que la palabra “precipitación” se refiere a extraer una sustancia específica de una solución. La proteína diana puede ser endógena o recombinante. La mayoría de las proteínas recombinantes tienen una etiqueta de epítopo (es decir, mico o bandera) unida a ellas para simplificar la purificación posterior. Típicamente, es más fácil optimizar la proteína recombinante IP porque los anticuerpos contra las etiquetas de epítopos recombinantes son muy fuertes y eficaces. Los anticuerpos contra las proteínas endógenas tienen una eficacia extremadamente variable, lo que hace mucho más difícil optimizar estas IP. Un paso necesario después de la inmunoprecipitación es la verificación de la purificación. La proteína aislada se resuelve utilizando SDS-PAGE y posteriormente sondeada para la pureza por manchas occidentales (Figura 1). Un control importante es el uso de un anticuerpo diferente durante la mancha occidental para verificar el tirón de la proteína correcta. La combinación de IP con técnicas posteriores es una poderosa herramienta de análisis. El objetivo después de la purificación puede ser la caracterización de la proteína en sí por RMN, espectrometría de masas y ensayos in vitro, o el análisis de los socios que interactúan con la proteína (es decir, proteína, ADN, ARN) (3, 4, 5).
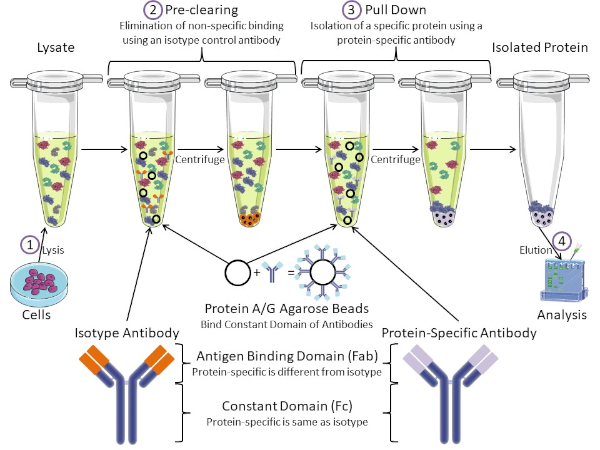
Figura 1: Visión general del procedimiento de inmunoprecipitación. La inmunoprecipitación es el aislamiento de una proteína específica utilizando un anticuerpo. Después de la producción de lisado a partir de células, hay dos pasos principales: pre-clearing y pull down. Durante el paso de pre-limpieza, los lisados celulares se eliminan previamente de proteínas que se unen a los anticuerpos no específicamente utilizando un anticuerpo de control de isotipo. En el paso de tracción hacia abajo, la proteína diana se tira hacia abajo usando un anticuerpo específico de la proteína. La proteína aislada es entonces analizada por western blot. Los anticuerpos isotipo y los anticuerpos específicos de proteínas tienen el mismo dominio constante, pero diferentes dominios de unión de antígenos. Un componente clave de este protocolo son las perlas de agarosa de proteína A/G que unen el dominio constante de los anticuerpos, lo que permite la inmunoprecipitación de la proteína diana. Haga clic aquí para ver una versión más grande de esta figura.
Los anticuerpos son el componente clave de una inmunoprecipitación que la diferencia de otras formas de purificación de proteínas (es decir, purificación de columnas de afinidad de níquel). Los anticuerpos son moléculas producidas por células B que pueden reconocer epítopos proteicos específicos. Los anticuerpos tienen dos dominios: constante (Fc) y enlace de antígeno (Fab) (Figura 1). El dominio constante identifica el tipo de anticuerpo y dicta la función in vivo. Por lo general, los dominios constantes de los anticuerpos utilizados para IP son IgG de ratón, rata o conejo. La porción de unión al antígeno del anticuerpo reconoce un epítopo específico de una proteína específica. Los anticuerpos pueden reconocer epítopos en proteínas plegadas que pueden no existir cuando la proteína está desnaturalizada y viceversa. Por lo tanto, la disponibilidad del epítopo depende del plegado de proteínas, identificando un factor importante a tener en cuenta a la hora de elegir anticuerpos y condiciones para la P.I.
Tanto los sistemas procaario como el eucariota tienen proteínas de unión a anticuerpos. En los sistemas eucariotas, el propósito es la protección inmune contra las bacterias mientras que en los sistemas procariotas, el propósito es la protección del sistema inmunológico. Las proteínas de unión a anticuerpos afectan a la metodología IP de dos maneras. En primer lugar, hay un paso necesario de pre-limpieza (Figura 1) para eliminar el lisado de proteínas que unen anticuerpos – reduciendo así la unión no específica en el producto final. Este paso utiliza un anticuerpo isotipo que tiene el mismo dominio constante que un dominio de unión de anticuerpos diferente al de su anticuerpo específico de proteína. Las proteínas bacterianas de unión a anticuerpos son el segundo componente clave de este método. Después de que el anticuerpo específico de la proteína une la proteína diana, el anticuerpo: complejo proteico debe ser arrastrado hacia abajo (Figura 1). Las proteínas A, G y L son proteínas bacterianas que unen el dominio constante de los anticuerpos. Mientras que las bacterias utilizan esto para subvertir el sistema inmunológico, los investigadores han cooptado este sistema para una fácil purificación de anticuerpos, y se utiliza durante los pasos previos a la limpieza y el tirón. Estas proteínas tienen diferentes afinidades de unión para diferentes especies y diferentes subtipos de dominio constante – otro factor a tener en cuenta a la hora de elegir condiciones para la P.I. Muchas empresas venden cuentas de agarosa etiquetadas con proteína A/G (Figura 1), columnas de espín prefabricadas o resinas para hacer columnas. En general, las perlas y las columnas de espín se utilizan para tamaños de muestra más pequeños, mientras que las resinas se utilizan para la purificación a granel.
En este ejercicio de laboratorio, demostramos cómo purificar la proteína endógena c-myc, a partir de timocitos murinoprimarios primarios, utilizando la técnica básica de inmunoprecipitación basada en la proteína A/G Plus. El protocolo comienza a partir de la preparación del lisado celular y termina con la verificación de la extracción exitosa de proteína hacia abajo usando el análisis de la mancha occidental.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.
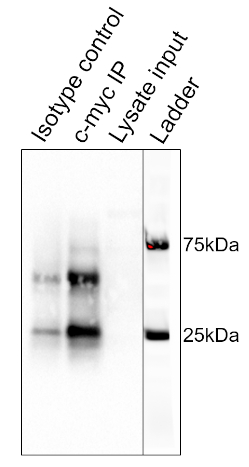
Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.