Fonte: Susannah C. Shissler1, Tonya J. Webb1
1 Dipartimento di Microbiologia e Immunologia, Università del Maryland, Baltimora, MD 21201
L’immunoprecipitazione (IP, noto anche come test “pull-down”) è una tecnica ampiamente utilizzata che ha applicazioni in una varietà di campi. Concepito per la prima volta nel 1984, è stato perfezionato nel 1988 (1, 2). L’obiettivo fondamentale dell’IP è la purificazione e l’isolamento di una proteina specifica utilizzando un anticorpo contro quella proteina. La parola “immuno” si riferisce all’uso di un anticorpo mentre la parola “precipitazione” si riferisce all’estrazione di una sostanza specifica da una soluzione. La proteina bersaglio potrebbe essere endogena o ricombinante. La maggior parte delle proteine ricombinanti ha un tag epitopo (cioè myc o bandiera) attaccato a loro per semplificare la successiva purificazione. In genere, è più facile ottimizzare la proteina IP ricombinante perché gli anticorpi contro i tag epitopi ricombinanti sono molto forti ed efficaci. Gli anticorpi contro le proteine endogene hanno un’efficacia estremamente variabile, rendendo molto più difficile ottimizzare queste IP. Un passo necessario dopo l’immunoprecipitazione è la verifica della purificazione. La proteina isolata viene risolta utilizzando SDS-PAGE e successivamente sonde per la purezza da western blots (Figura 1). Un controllo importante è l’uso di un anticorpo diverso durante il Western blot per verificare il pull down della proteina corretta. La combinazione di IP con tecniche successive è un potente strumento di analisi. L’obiettivo dopo la purificazione può essere la caratterizzazione della proteina stessa mediante NMR, spettrometria di massa e saggi in vitro, o l’analisi dei partner interagenti della proteina (cioè proteine, DNA, RNA) (3, 4, 5).
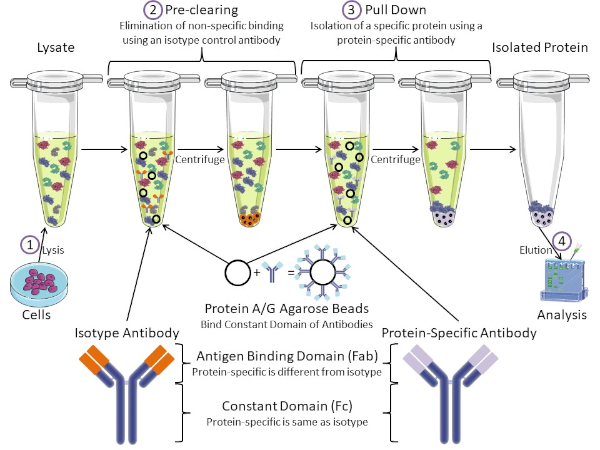
Figura 1: Panoramica della procedura di immunoprecipitazione. L’immunoprecipitazione è l’isolamento di una proteina specifica utilizzando un anticorpo. Dopo la produzione di lisi dalle cellule, ci sono due passaggi principali: pre-pulizia e pull down. Durante la fase di pre-clearing, i lasati cellulari viene pre-eliminata dalle proteine che si legano agli anticorpi in modo non specifico utilizzando un anticorpo di controllo dell’isotipo. Nella fase di pull down, la proteina bersaglio viene tirata verso il basso usando un anticorpo specifico della proteina. La proteina isolata viene quindi analizzata da Western blot. Gli anticorpi isotipia e gli anticorpi specifici della proteina hanno lo stesso dominio costante, ma domini di legame dell’antigene diversi. Un componente chiave di questo protocollo sono le perle di agarose della proteina A / G che legano il dominio costante degli anticorpi, consentendo l’immunoprecipitazione della proteina bersaglio. Fare clic qui per visualizzare una versione più grande di questa figura.
Gli anticorpi sono il componente chiave di un’immunoprecipitazione che la differenziano da altre forme di purificazione delle proteine (cioè la purificazione della colonna di affinità del nichel). Gli anticorpi sono molecole prodotte da cellule B in grado di riconoscere specifici epitopi proteici. Gli anticorpi hanno due domini: costante (Fc) e legame con l’antigene (Fab) (Figura 1). Il dominio costante identifica il tipo di anticorpo e detta la funzione in vivo. Di solito, i domini costanti degli anticorpi utilizzati per l’IP sono IgG di topo, ratto o coniglio. La porzione di legame con l’antigene dell’anticorpo riconosce un epitopo specifico di una proteina specifica. Gli anticorpi possono riconoscere gli epitopi su proteine ripiegate che potrebbero non esistere quando la proteina è denaturata e viceversa. Pertanto, la disponibilità dell’epitopo dipende dal ripiegamento delle proteine, identificando un fattore importante da considerare quando si scelgono anticorpi e condizioni per l’IP.
Sia il sistema procariotico che eucariotico hanno proteine leganti gli anticorpi. Nei sistemi eucariotici, lo scopo è la protezione immunitaria dai batteri mentre nei sistemi procariotici, lo scopo è la protezione dal sistema immunitario. Le proteine leganti gli anticorpi influenzano la metodologia IP in due modi. In primo luogo, c’è una fase di pre-clearing necessaria (Figura 1) per liberare il lisiato dalle proteine che legano gli anticorpi, riducendo così il legame non specifico nel prodotto finale. Questo passaggio utilizza un anticorpo isotipo che ha lo stesso dominio costante ma un dominio di legame anticorpale diverso rispetto all’anticorpo specifico della proteina. Le proteine che legano gli anticorpi batterici sono il secondo componente chiave di questo metodo. Dopo che l’anticorpo proteina-specifica lega la proteina bersaglio, il complesso anticorpo: proteina deve essere tirato verso il basso (Figura 1). Le proteine A, G e L sono proteine batteriche che legano il dominio costante degli anticorpi. Mentre i batteri usano questo per sovvertire il sistema immunitario, i ricercatori hanno cooptato questo sistema per una facile purificazione degli anticorpi, e viene utilizzato sia durante le fasi di pre-clearing che di pull-down. Queste proteine hanno diverse affinità di legame per diverse specie e diversi sottotipi di dominio costante – un altro fattore da considerare quando si scelgono le condizioni per IP. Molte aziende vendono perle di agarose marcate Protein A / G (Figura 1), colonne di spin prefabbricate o resine per realizzare colonne. In generale, le pere e le colonne di spin vengono utilizzate per campioni di dimensioni inferiori, mentre le resine vengono utilizzate per la purificazione alla rinfusa.
In questo esercizio di laboratorio, dimostriamo come purificare la proteina endogena c-myc, dai timociti murini primari, utilizzando la tecnica di immunoprecipitazione di base basata sulle perle di agarose Protein A / G Plus. Il protocollo inizia dalla preparazione del lizzata cellulare e termina con la verifica del successo del pull down proteico utilizzando l’analisi Western blot.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.
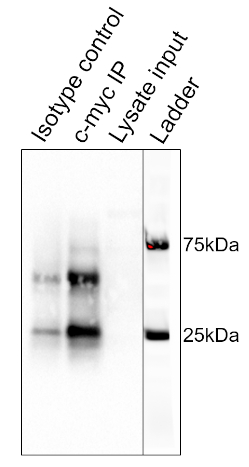
Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.