A series of bsRNA-seq libraries from cell lines19 were generated by following the procedures in this report (Figure 1). After total RNA purification accompanied by DNase treatment performed on cell line samples and the quality checked by gel electrophoresis and UV-Vis spectrophotometry (A260/A280), the RNA sample can proceed to poly(A) RNA enrichment. To determine whether the double purification could remove the majority of ribosomal RNA, the purification efficiency of poly(A) RNA was assessed by capillary electrophoresis total RNA assay that can automatically calculate the rRNA contamination percentage (Figure 2). From the RNA fragment peak pattern and rRNA contamination percentage, RNA samples with twice purification indeed showed decreased contamination of ribosomal RNA from 6.5% to 2.2% and 2.6% to 1.1% in AsPC-1 and BxPC-3, respectively. With human pancreatic cancer cell lines, two rounds of beads purification could reach an average of 2.8 ± 1% poly(A)-enriched RNA abundances from the total RNA samples. Therefore, the poly(A) RNA enrichment by oligo(dT) beads with double purification steps minimized ribosomal RNA and represents a feasible mRNA sample enrichment for the downstream experiments.
The bisulfite conversion protocols had been reported in several studies on RNA 5-methylcytosine modification (Table 1). The bisulfite reaction could be performed with a user-reconstituted reaction mixture consisting of 40% sodium bisulfite and 600 mM hydroquinone in the aqueous solution (pH 5.1) at 75 °C for 4 hours10,20,21. Alternatively, a more stringent bisulfite treatment of RNA sample may also be performed with a commercial kit; which in a number of studies from others and ours6,22,23,24 extended the bisulfite reaction time to three incubation cycles. Since the three-cycle incubation step efficiently converts unmethylated cytosine in the in vitro transcribed and structurally more-folded Escherichia coli 16S rRNA segment8, the three-cycle incubation step was also applied in this protocol.
With twice poly(A) enrichment and the three-cycle bisulfite conversion, RNA quality before and after the reaction was assessed by capillary electrophoresis. The RNA size distribution after the bisulfite treatment showed a peak of ~200-500 nucleotides because of fragmentation caused by the bisulfite reaction (Figure 3). Additionally, the fragmented bsRNA is ideal for library preparation without the need to conduct another fragmentation step. A total of 8-10 ng of bsRNA as input was used to construct the library according to this protocol by using 9 to 11 cycles in the final PCR amplification of adapter-ligated DNA. The capillary electrophoresis of the amplicon showed quite a successful library preparation, with only a small portion of primer remaining and no peak of over-amplification (Figure 4). To check the conversion efficiency in each sample library, the unmethylated firefly luciferase RNA as a spike-in control sequence was added to the sample before conducting the bisulfite treatment. After the sequencing reads aligned to the reference sequence using meRanTK tool kits25, the average of 2,440 (0.006%) of total reads were mapped to the spike-in sequence, and the total analyzed C-to-T conversion rate reached an average of 99.81%; the status can be viewed in Integrated Genomics Viewer (Figure 5).
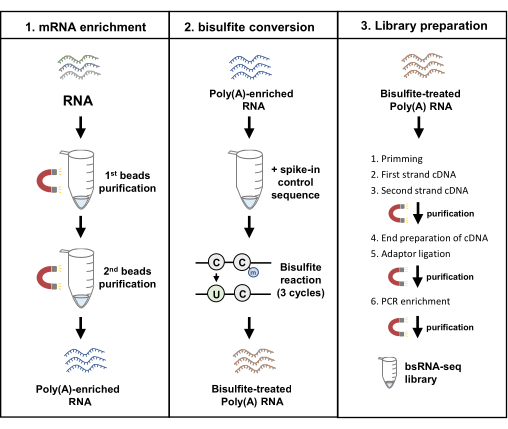
Figure 1: Workflow diagram of bisulfite mRNA sequencing. The pipeline constitutes of three main protocols, including mRNA enrichment, bisulfite conversion, and library preparation. Abbreviation: bsRNA = bisulfite mRNA; QC. = quality control. Please click here to view a larger version of this figure.
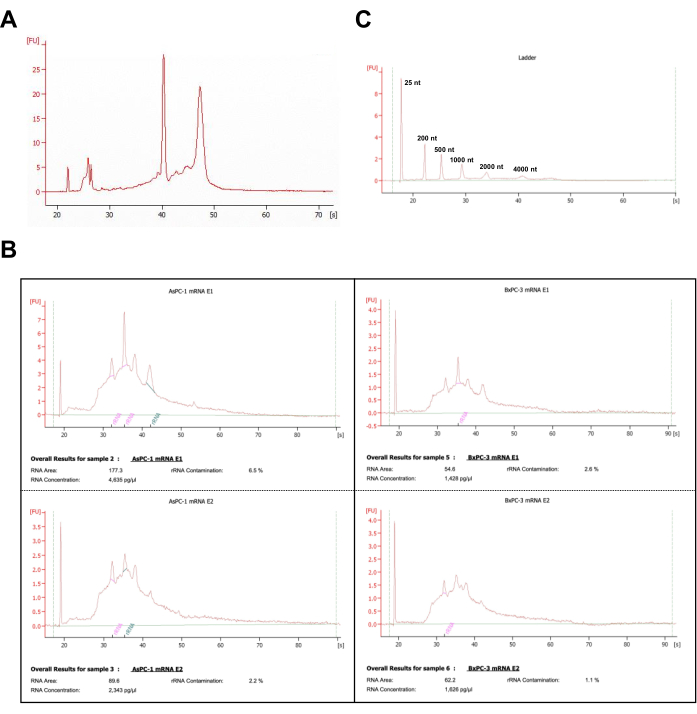
Figure 2: Quality control results of poly(A)-enriched RNA samples with capillary electrophoresis. (A) The representative capillary electrophoresis profile of the total RNA from BxPC-3 cells. (B) The representative capillary electrophoresis profiles of samples processed with oligo(dT) purification from AsPC-1 and BxPC-3 cells. mRNA E1, mRNA elution from one-time purification; mRNA E2, mRNA elution from two-times purification. The rRNA contamination estimates were calculated by the electrophoresis operation system that controls the total RNA analysis assay. (C) The capillary electrophoresis profile of the Ladder markers was obtained in the same run with samples presented in panel B. Please click here to view a larger version of this figure.
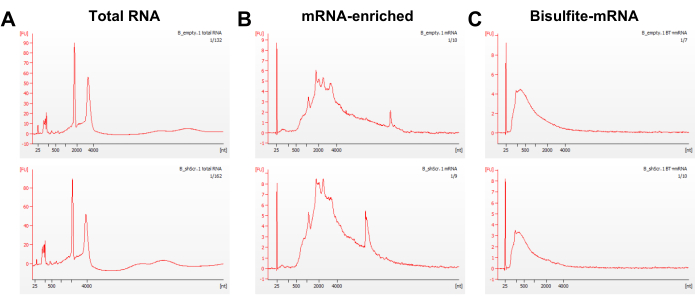
Figure 3: Quality assessment of RNA samples by capillary electrophoresis. The representative capillary electrophoresis profiles of (A) the total RNA, (B) the poly(A)-enriched mRNA from two times purification, and (C) the bisulfite-treated mRNA from the BxPC-3 cells. Abbreviations: B_empty = BxPC-3 cells transduced with empty vector; B_shScr = BxPC-3 cells transduced with the scramble sequences. Please click here to view a larger version of this figure.
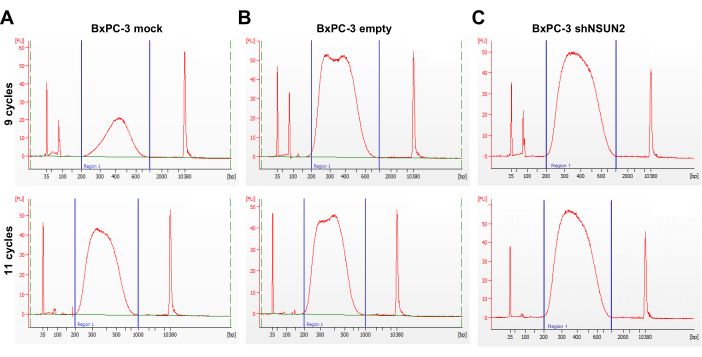
Figure 4: Quality assessment of bsRNA-seq libraries. (A-C) Three sets of representative capillary electrophoresis profiles of constructed bsRNA-seq libraries amplified by PCR at different PCR cycle numbers of 9 and 11 cycles using bsRNA from a set of BxPC-3 cells. The estimated input quantity of the bisulfite-treated RNA was determined by a high-sensitivity RNA quantification assay. A total of 8, 8.4, and 9.6 ng bsRNA were used in the (A) BxPC-3 mock, (B) BxPC-3 empty, and (C) BxPC-3 shN2UN2 samples, respectively. The ladder marker peaks at 35 bp and the 10,380 bp represent the internal standards of the lower- and upper-boundary in the electrophoresis profile of each sample. A prominent peak of around 100 bp and an obscure peak of around 127 bp, respectively, indicate the PCR primers (<100 bp) and the adaptor-dimer (~127 bp), which remained in the eluate after PCR cleanup. Abbreviations: BxPC-3 mock = untransduced BxPC-3 cells; BxPC-3 empty = BxPC-3 cells transduced with the empty vector control; BxPC-3 shN2UN2 = BxPC-3 cells transduced with the shNSUN2 constructs. Please click here to view a larger version of this figure.
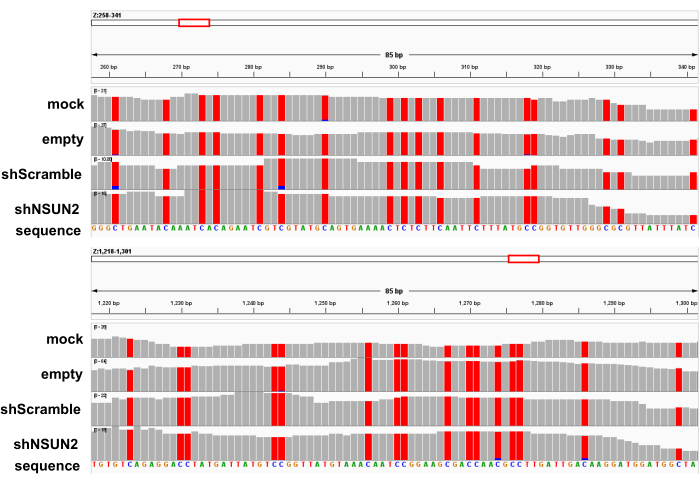
Figure 5: The sequence alignment profiles of each bsRNA-seq library mapped to the spike-in-control reference sequence. The representative alignment profiles of the BxPC-3 mock, empty, scramble, and shNSUN2 bsRNA-seq libraries to the firefly luciferase spike-in control reference sequence (indicated as the "Z" gene); 2 representative regions were shown. Grey bars indicate all reads that present matched consensus nucleotides at the indicated base position to the reference sequence. Red bars highlight the thymidine (T) identity at the base position of the reads; blue bars indicate those reads having the cytosine (C) identity at the position. Please click here to view a larger version of this figure.
Table 1: Comparison of bisulfite reaction protocols and the conversion rate. Please click here to download this Table.
| Agilent 2100 Electrophoresis Bioanalyzer System | Agilent, Santa Clara, CA | RNA quality detection | |
| AMpure XP beads | Beckman Coulter | A63881 | purify DNA |
| Bioanalyzer DNA high sensitivity kit | Agilent, Santa Clara, CA | 5067-4626 | DNA quality dection |
| Bioanalyzer RNA 6000 Pico kit | Agilent, Santa Clara, CA | 5067-1513 | RNA quality dection |
| DiaMag02 – magnetic rack | Diagenode, Denville, NJ | B04000001 | assist library preparation |
| DiaMag1.5 – magnetic rack | Diagenode, Denville, NJ | B04000003 | assist poly(A) RNA purificaion |
| Dynabeads mRNA DIRECT purification kit | Thermo Fisher Scientific, Waltham, MA | 61011 | poly(A) RNA purificaion; Wash Buffer 1 and Wash Buffer 2 |
| Ethanol | J.T.Baker | 64-17-5 | |
| EZ RNA methylation kit | Zymo, Irvine, CA | R5002 | bisulfite treatment |
| Firefly luciferase mRNA | Promega, Madison, WI, USA | L4561 | spike in control seqeunce |
| KAPA Library Quantification Kits | Roche, Switzerland | KK4824 | library quantification |
| Nanodrop spectrophotometer | Thermo Fisher Scientific, Waltham, MA | Total RNA quantity detection | |
| NEBNext multiplex Oligos for illumina (index Primer set1) | New England Biolabs, Ipswich, MA | E7335S | library preparation |
NEBNext Ultra  Directional RNA Library Prep Kit for Illumina Directional RNA Library Prep Kit for Illumina |
New England Biolabs, Ipswich, MA | E7760S | library preparation |
| Nuclease-free Water | Thermo Fisher Scientific | AM9932 | |
| P2 pipetman | Thermo Fisher Scientific, Waltham, MA | 4641010 | |
| Qubit 2.0 fluorometer | Thermo Fisher Scientific, Waltham, MA | RNA quantity detection | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific, Waltham, MA | Q32854 | DNA quantity detection |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific, Waltham, MA | Q32852 | RNA quantity detection |
