La nostra comprensione della differenziazione cellulare e della genesi di tessuti e organi è il risultato di decenni di elaborati screening mirati di geni e dei loro prodotti. Aumentare la nostra conoscenza di tutte le biomolecole e delle loro quantità durante importanti eventi cellulari aiuterebbe a svelare i meccanismi molecolari che controllano il modello spaziale e temporale del piano corporeo dei vertebrati. Le tecnologie che consentono l’amplificazione molecolare e il sequenziamento sono ora in grado di riferire regolarmente su un gran numero di geni e trascrizioni, supportando studi basati su ipotesi nella ricerca biologica e traslazionale di base. Per comprendere i sistemi in via di sviluppo, una complessa relazione tra trascrizione e traduzione sostiene l’analisi diretta di più proteine e le loro modificazioni post-traduzionali. La proteomica globale che utilizza sistemi biologici in vitro, come le cellule staminali pluripotenti indotte, ha iniziato a delineare i meccanismi di induzione tissutale 1,2. Negli organismi complessi, come l’embrione vertebrato, lo sviluppo si basa su gradienti morfogeni nel contesto dello spazio e del tempo3. Ne consegue che acquisire conoscenze sui cambiamenti proteomici man mano che le cellule si differenziano per formare tessuti specializzati, come i tessuti neurali, offre una chiave per sbloccare programmi molecolari che controllano lo sviluppo normale e difettoso e guidare le terapie di prossima generazione.
La rana artigliata sudafricana vertebrata (Xenopus laevis) è un modello consolidato nella biologia cellulare e dello sviluppo, neuro e rigenerativa. Il Premio Nobel 2012 per la Fisiologia ola Medicina 4,5 di Sir John Gurdon per la scoperta della pluripotenza del nucleo somatico ha evidenziato l’importanza di questo modello per le scoperte negli studi di base e traslazionali. Gli embrioni di Xenopus si sviluppano esternamente alla madre, facilitando così la manipolazione diretta delle cellule, dei cloni cellulari e dell’espressione genica nelle varie fasi di sviluppo. La pigmentazione asimmetrica e le divisioni cellulari stereotipate hanno permesso di tracciare mappe del destino riproducibili dall’embrione a 7,8 e 16-6e 32 cellule. Per la proteomica basata sulla spettrometria di massa ad alta risoluzione (HRMS), ulteriori vantaggi del modello includono dimensioni relativamente grandi (~ 1 mm di diametro), che producono un abbondante contenuto proteico per l’analisi (~ 130 μg negli embrioni in fase di scissione precoce, ~ 10 μg di contenuto proteico in singole cellule dell’embrione a 16 cellule)9,10.
Attualmente, HRMS è la tecnologia leader di scelta per la rilevazione delle proteine. Questa tecnologia consente il rilevamento e la quantificazione diretti, sensibili e specifici di proteine multiple, di solito da centinaia a migliaia di proteine diverse11. La proteomica bottom-up di HRMS comporta una serie di passaggi interconnessi. Dopo l’estrazione dal campione di cellula/tessuto, le proteine vengono digerite con un enzima proteolitico, come la tripsina (proteomica bottom-up). I peptidi risultanti vengono separati in base alle loro diverse proprietà fisico-chimiche, tra cui idrofobicità (cromatografia liquida a fase inversa, LC), carica netta (cromatografia a scambio ionico), dimensioni (cromatografia di esclusione dimensionale) o mobilità elettroforetica (elettroforesi capillare, CE). I peptidi vengono quindi caricati (ionizzati), tipicamente utilizzando la ionizzazione elettrospray (ESI), e gli ioni peptide vengono rilevati e sequenziati tramite frammentazione in fase gassosa mediante HRMS tandem. I dati peptidici risultanti sono mappati sul proteoma dell’organismo studiato. Con l’intensità del segnale dello ione peptidico specifica per proteine (proteotipica) correlata alla concentrazione, la quantificazione delle proteine può essere eseguita senza etichetta o basata sull’etichetta (quantificazione multiplexing). La proteomica HRMS fornisce una ricca risorsa di informazioni sullo stato molecolare del sistema in studio, consentendo la generazione di ipotesi e studi funzionali di follow-up.
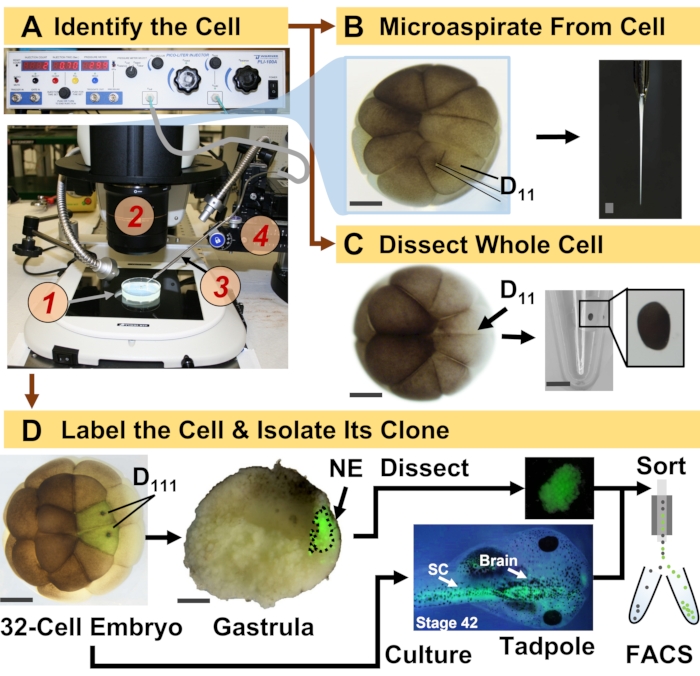
Figura 1: Proteomica spaziotemporalmente scalabile che consente la proteomica HRMS guidata dalla linea cellulare nell’embrione in via di sviluppo (rana). (A) Visualizzazione del campione (1) utilizzando uno stereomicroscopio (2) per l’iniezione di una cellula identificata (inserto), utilizzando una micropipetta fabbricata (3) sotto controllo da uno stadio di traslazione (4). (B) Campionamento subcellulare della cellula D11 sinistra identificata in un embrione a 16 cellule. (C) Dissezione di un’intera cellula D11 da un embrione a 16 cellule. (D) Tracciamento fluorescente (verde) delle progenie D111 sinistra e destra da un embrione a 32 cellule per guidare la dissezione dell’ectoderma neurale (NE) nella gastrula (stadio 10) e l’isolamento del tessuto discendente dal girino mediante FACS. Barre della scala: 200 μm per gli embrioni, 1,25 mm per la fiala. Le figure sono state adattate con il permesso dei riferimenti 15,19,21,59. Fare clic qui per visualizzare una versione ingrandita di questa figura.
Il protocollo qui presentato consente la quantificazione basata su HRMS di un gran numero di proteine in cellule/tessuti identificati nello sviluppo di embrioni di X. laevis. L’approccio si basa su un’accurata identificazione cellulare, mappe del destino cellulare riproducibili e metodologie consolidate per tracciare le linee cellulari in questo modello biologico 6,7,8. Come mostrato nella Figura 1, studiamo i proteomi da singole cellule impiegando la dissezione dell’intera cellula o il microcampionamento capillare per aspirare il contenuto cellulare. Il monitoraggio del lignaggio di una cellula ci permette di studiare l’evoluzione spaziotemporale del proteoma quando le cellule formano i tessuti durante la gastrulazione. La progenie cellulare è marcata in modo fluorescente iniettando un fluoroforo coniugato a destrano inerte o mRNA per proteine fluorescenti (ad esempio, proteina fluorescente verde o GFP). La progenie marcata è isolata nei punti temporali di sviluppo desiderati. Durante la gastrulazione, i cloni cellulari che sono strettamente raggruppati possono essere isolati per dissezione. Dopo la gastrulazione, i cloni cellulari possono essere distribuiti all’interno dell’embrione a causa di movimenti migratori e possono essere isolati dai tessuti dissociati mediante selezione cellulare attivata dalla fluorescenza (FACS). Le proteine in queste cellule e tessuti sono misurate tramite proteomica bottom-up che impiega HPLC o CE per la separazione e HRMS tandem ESI per l’identificazione. La proteomica HRMS guidata dalla linea cellulare è scalabile a diverse dimensioni cellulari e linee cellulari all’interno dell’embrione ed è specifica, sensibile e quantitativa. Attraverso esempi selezionati mostrati qui, dimostriamo anche che questo protocollo è scalabile e ampiamente adattabile a diversi tipi di cellule e linee cellulari.
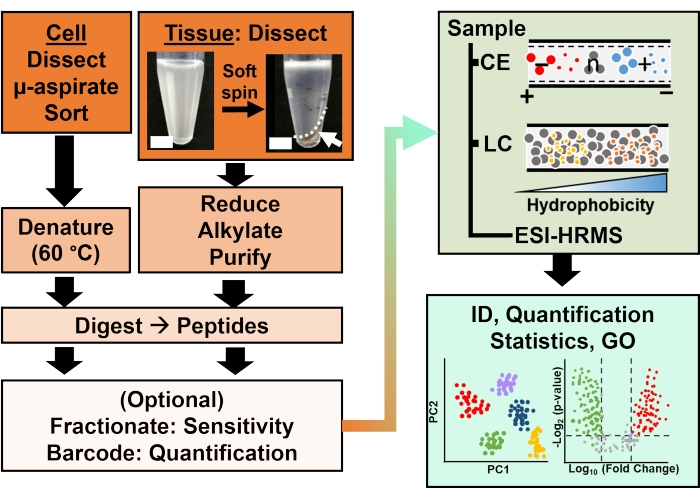
Figura 2: Il flusso di lavoro bioanalitico. Micro-dissezione e aspirazione capillare, o FACS ha facilitato il campionamento del contenuto proteico cellulare e clonale. Deplezione delle abbondanti proteine del tuorlo e separazione mediante elettroforesi capillare (CE) o cromatografia liquida a nanoflusso (LC) con sensibilità di identificazione migliorata (ID) mediante spettrometria di massa ad alta risoluzione (HRMS) a ionizzazione elettrospray (ESI). La quantificazione ha rivelato la disregolazione, fornendo nuove informazioni per studi basati su ipotesi in combinazione con le informazioni disponibili dall’ontologia genica (GO). Le cifre sono state adattate con il permesso del riferimento15. Fare clic qui per visualizzare una versione ingrandita di questa figura.
